计算 / 分析方法
吸附能(Adsorption energy)是表征分子与固体表面相互作用强度的关键物理量,反映吸附前后体系的能量变化。其数值直接决定吸附稳定性、覆盖度与吸附构型,是评估表面活性位点、预测吸附行为与催化潜力的核心参数。在第一性原理计算中,高精度吸附能可量化界面作用,为催化材料设计与反应机理研究提供可靠理论依据。
计算图谱
计算图谱与结果示例
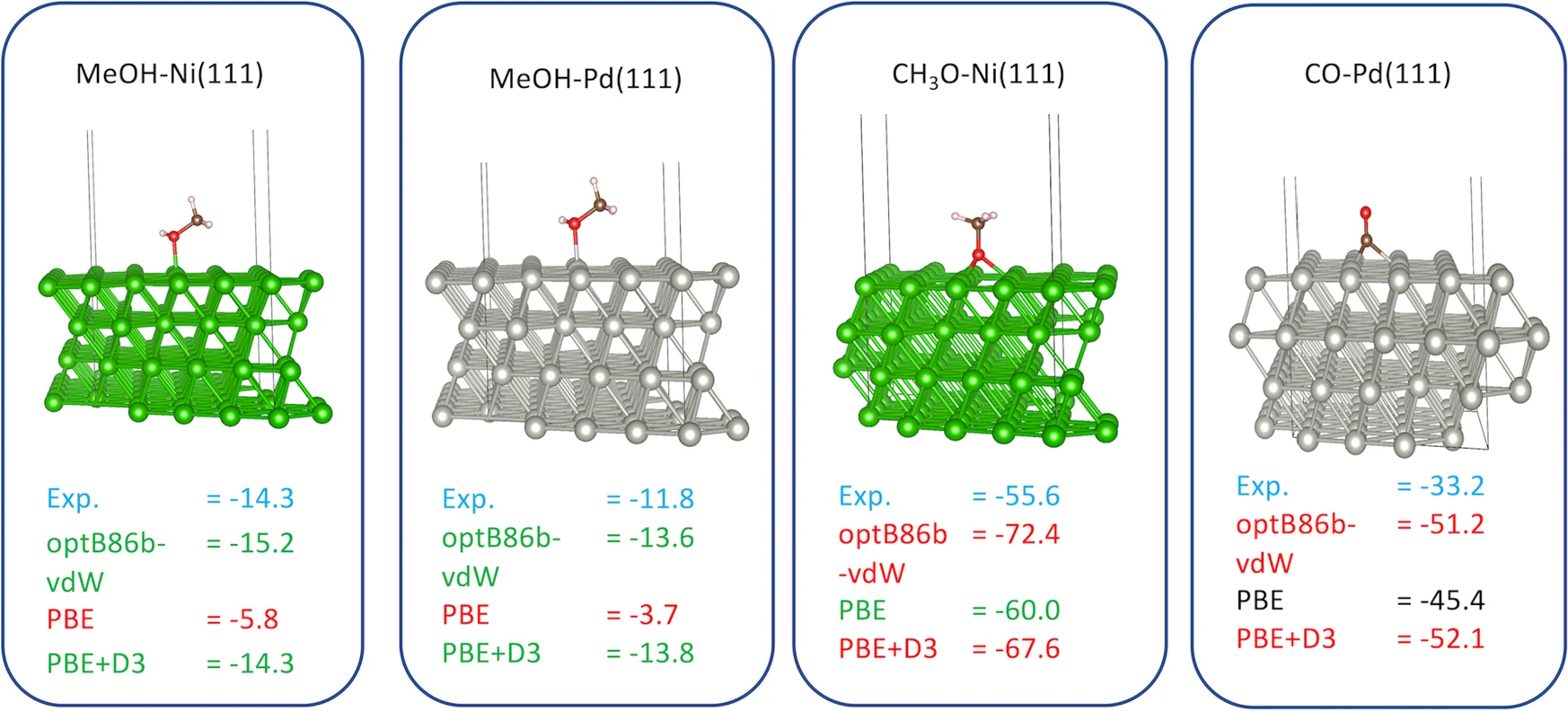
来自客户第一性原理材料“吸附能”对应页的计算图谱,用于说明吸附构型与能量比较的展示形态;不作为已交付项目成果承诺。
客户需提供材料
建议提供研究方向、结构/模型/数据文件、目标性质或指标、软件偏好、体系规模和交付周期。
交付结果
交付范围按项目确认,可包含输入输出文件、关键图谱、数据表、运行日志和分析说明。