典型任务
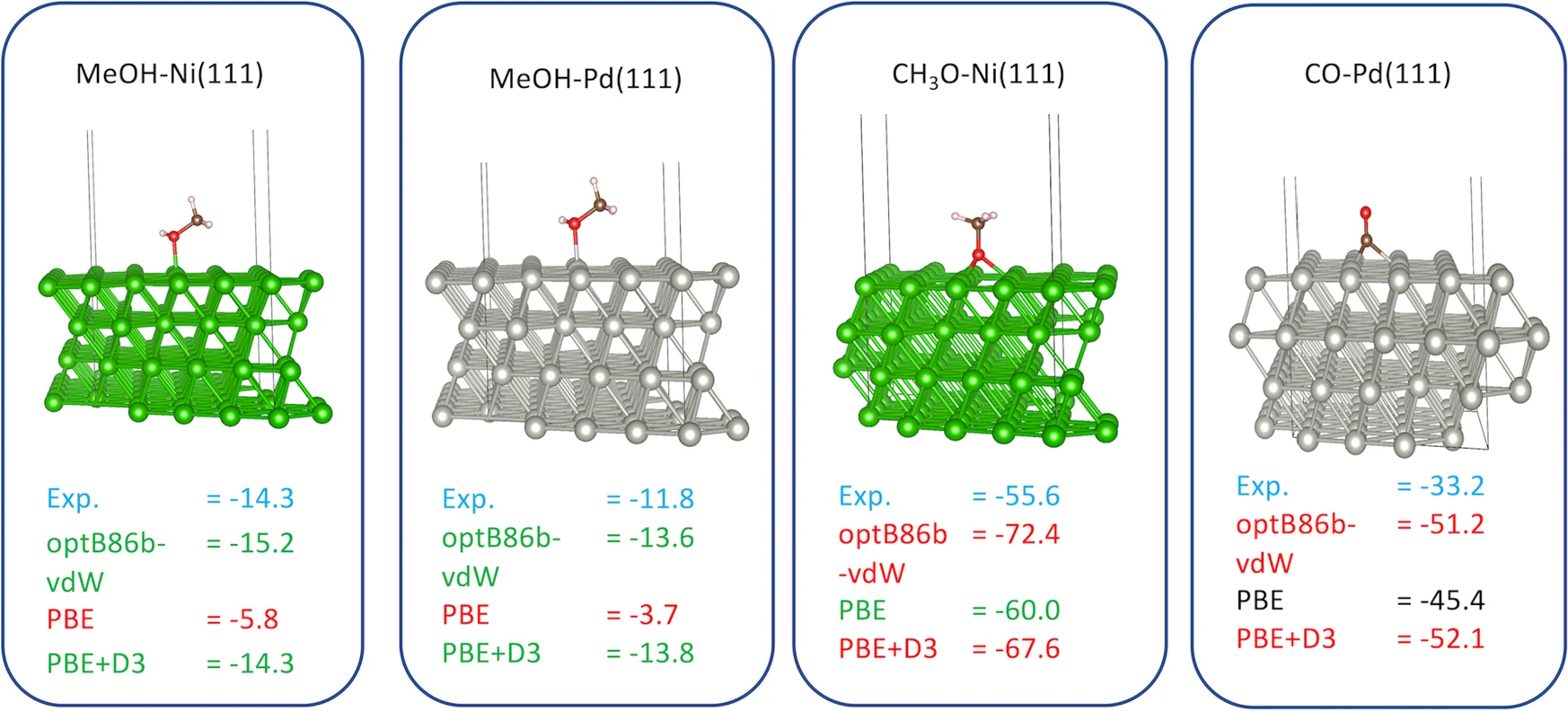
吸附能
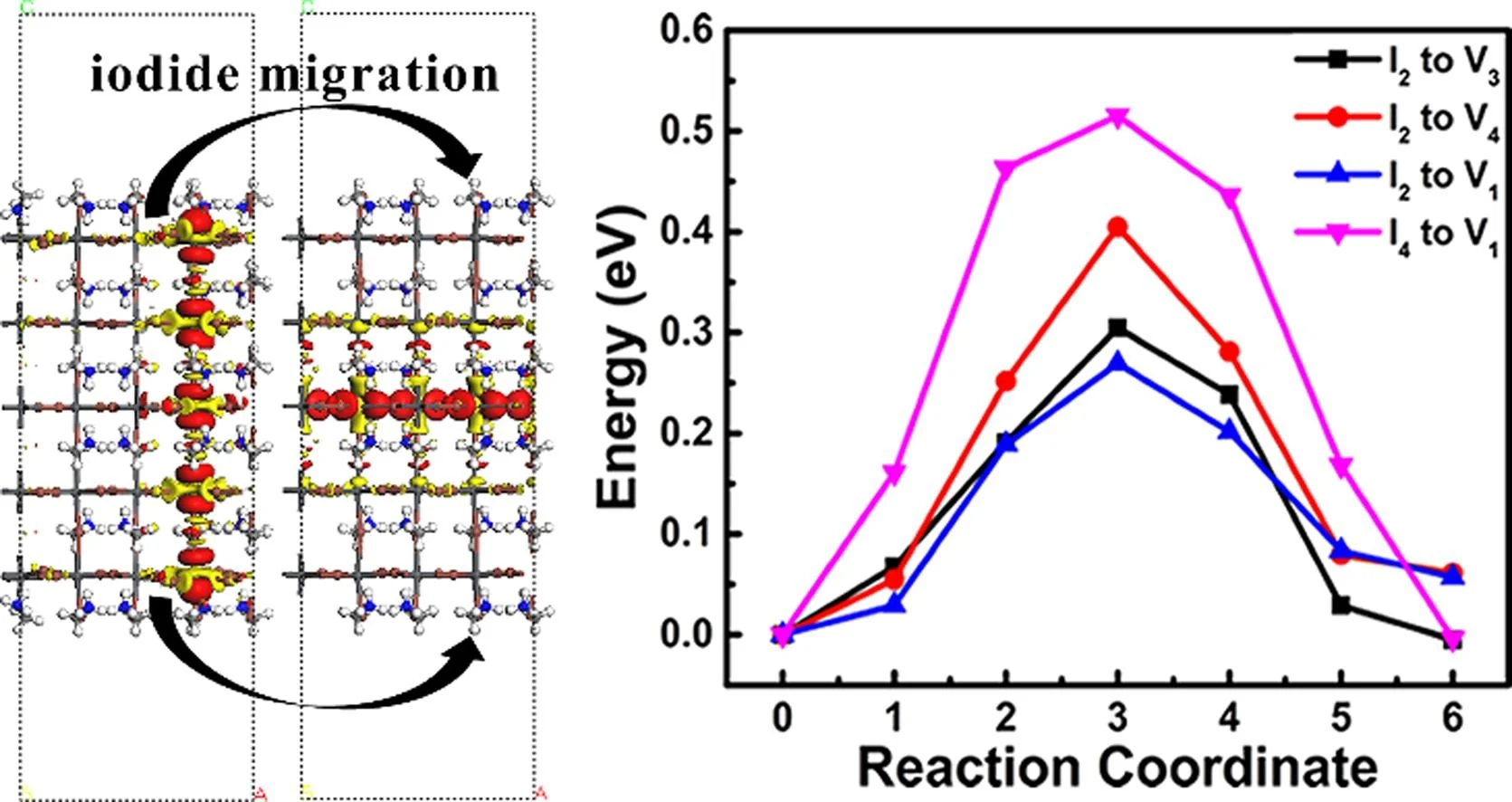
迁移能垒
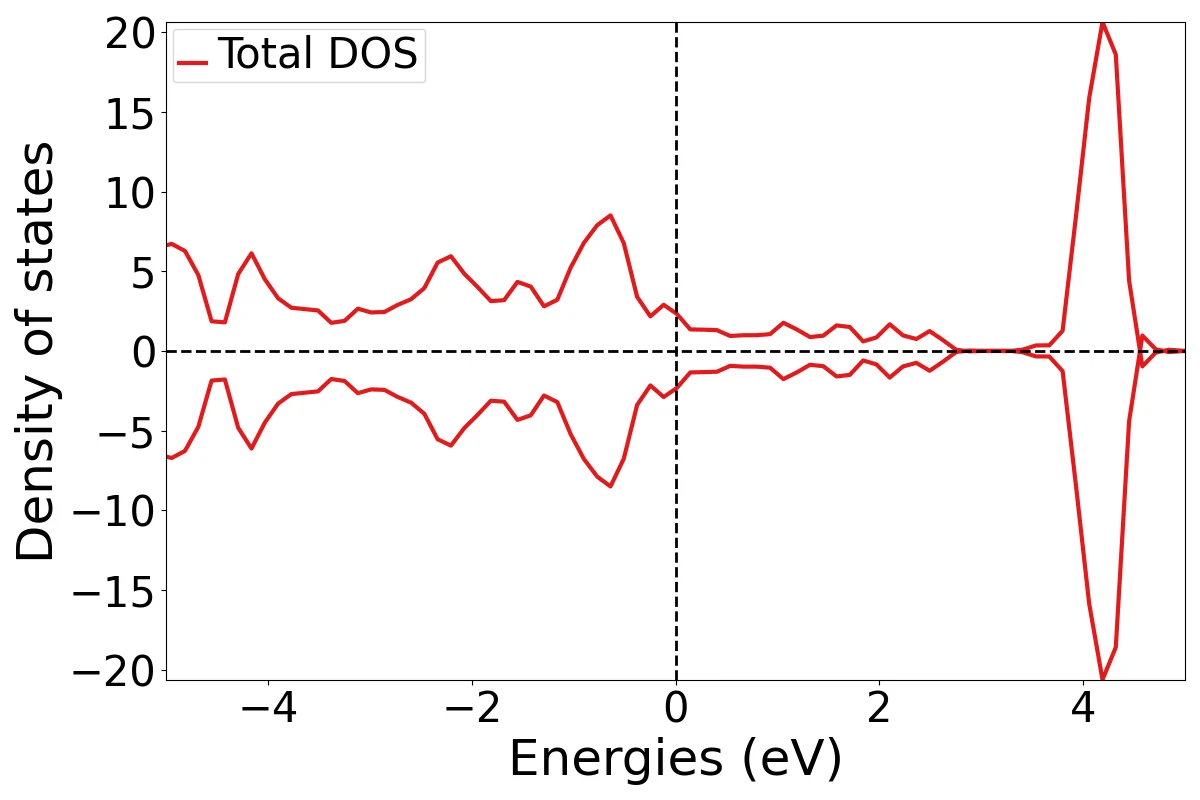
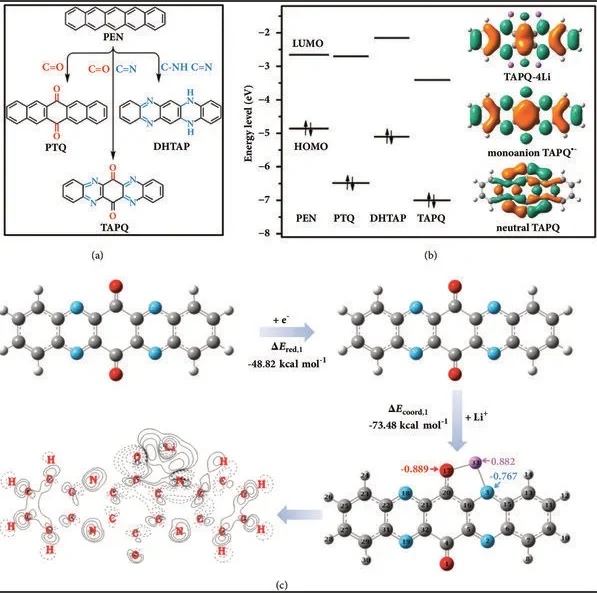
态密度
投影态密度(PDOS)
界面形成能
差分电荷
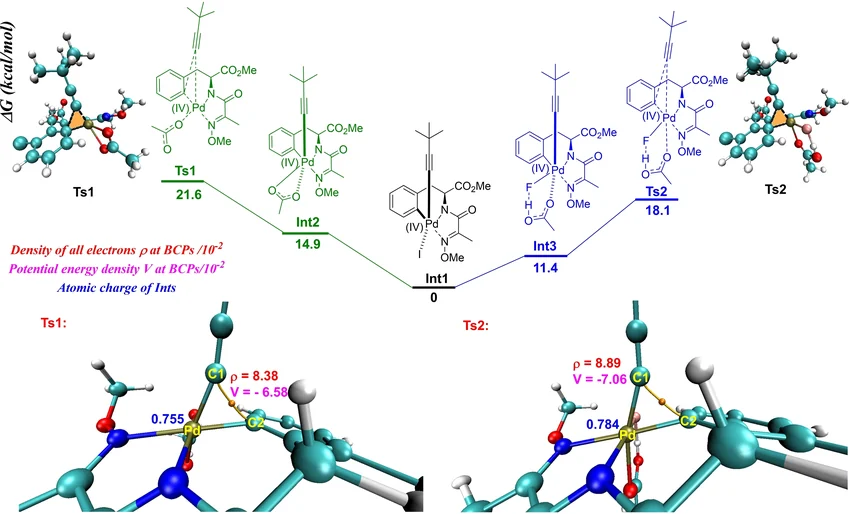
吉布斯自由能
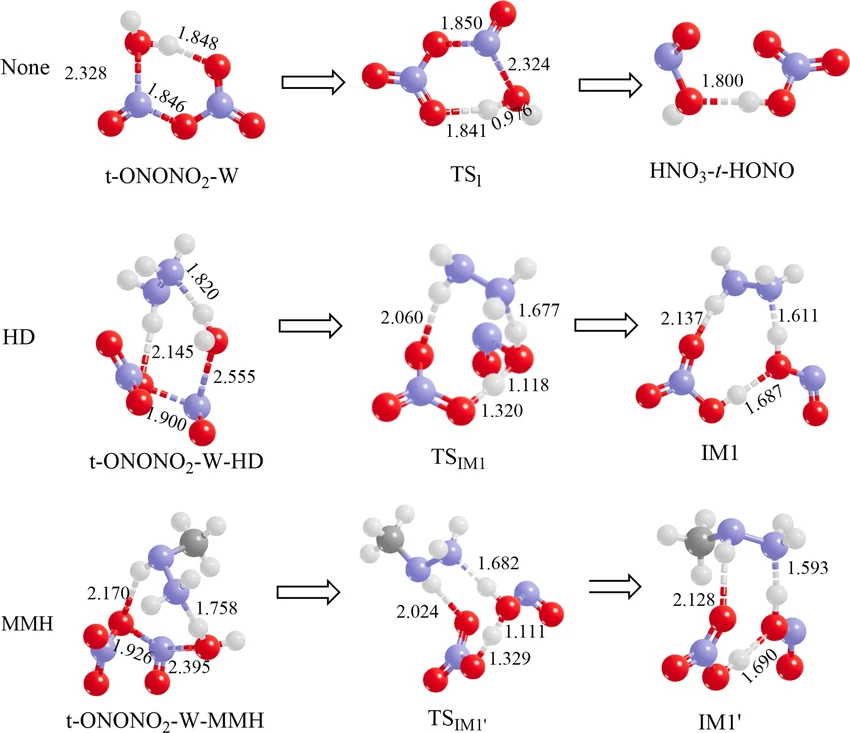
反应能垒
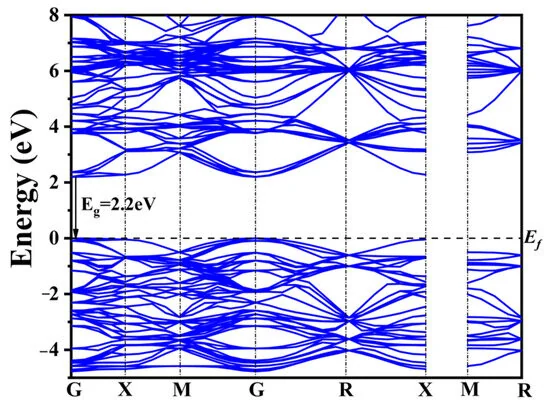
能带结构
反应路径
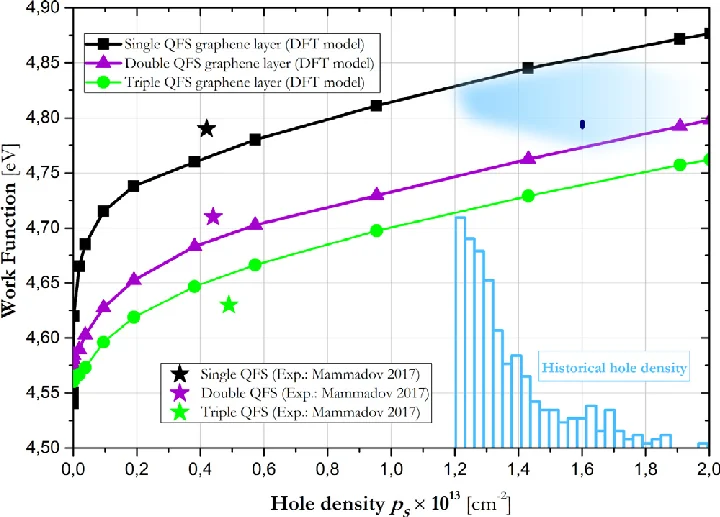
功函数
声子谱
费米能级
电荷局域密度
Bader电荷
介电常数
晶格常数
弹性模量
形成能
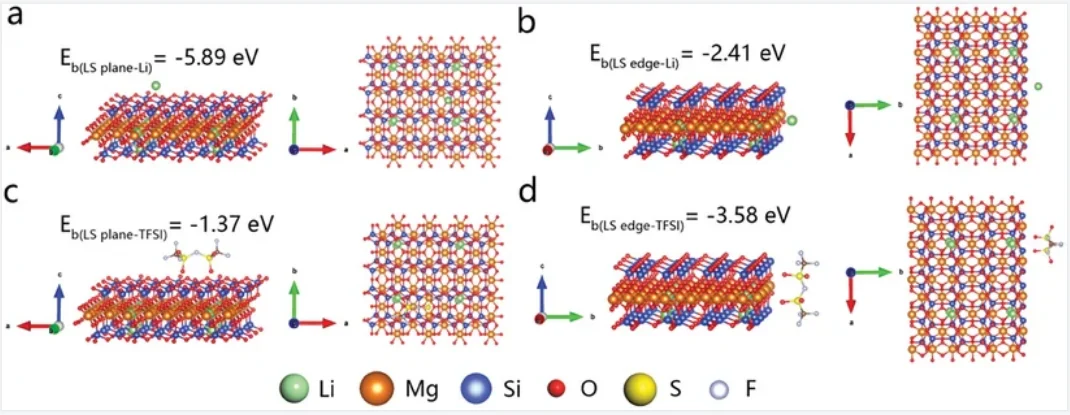
结合能
异质结
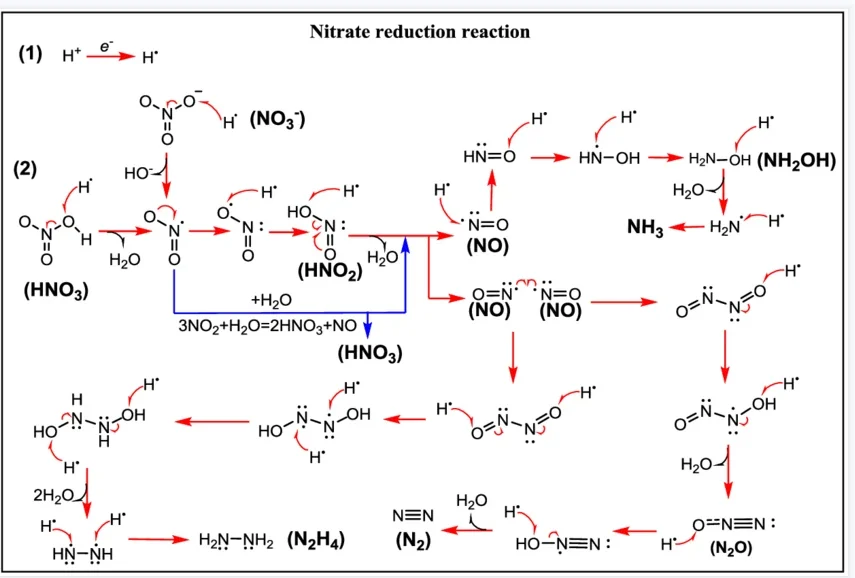
ORR(氧还原反应)
OER(电催化析氧反应)
HER(电催化析氢反应)
COHP(晶体轨道哈密顿布居)
AIMD(从头算分子动力学)
栏目内容
按任务条目展开的计算内容
每个条目都有独立 URL、SEO 和搜索命中,用户可以按任务名称、分析指标和软件方向快速进入对应内容。
04
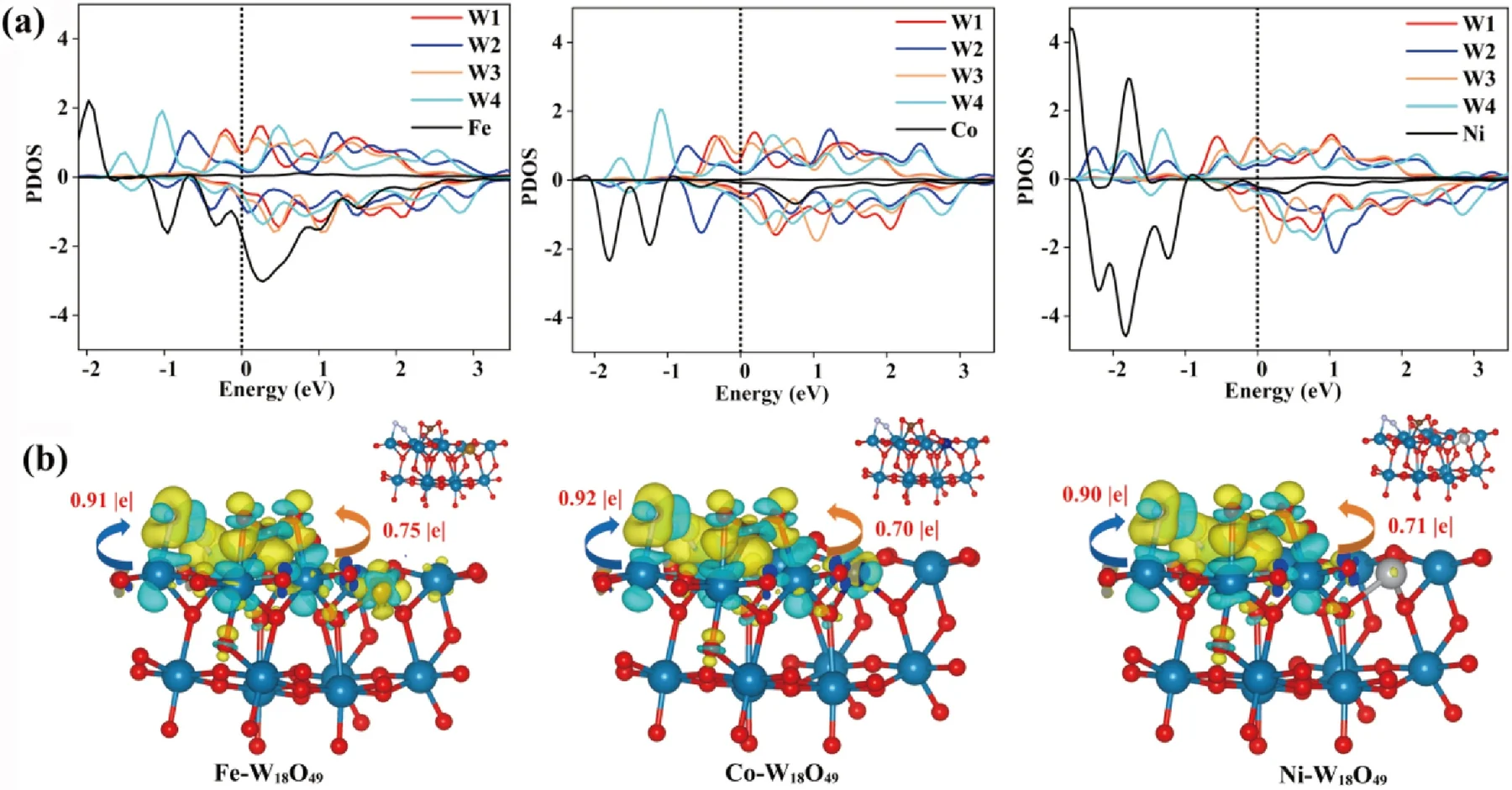
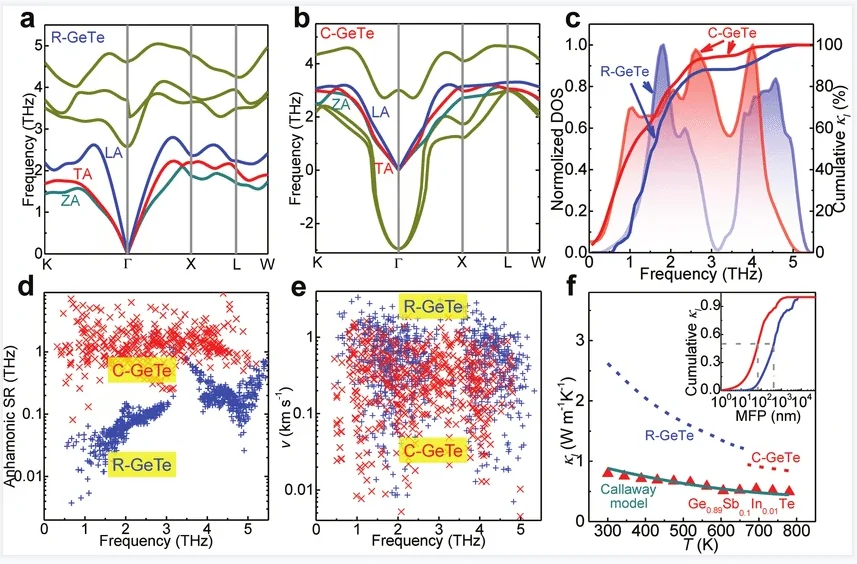
投影态密度
投影态密度(PDOS)
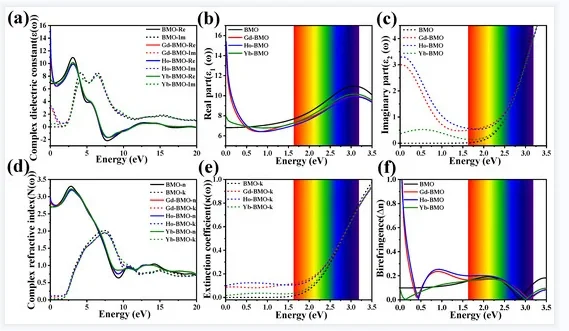
投影态密度(PDOS)是将总态密度按原子与轨道成分分解,定量表征各原子、各原子轨道对电子态的贡献。结合第一性原理计算,可直观解析费米能级附近轨道起源、电子结构与成键特征,为材料导电性、催化活性及能带机理提供精细的原子层面依据。
查看条目
22
ORR
ORR(氧还原反应)
ORR(氧还原反应)是能源催化领域核心反应,碱性条件下存在四电子(生成水,涉及 * OOH、*O、OH 中间体)与二电子(生成 H₂O₂,仅含OOH 中间体)两种路径。基于 DFT 计算可解析反应路径、理论过电位及自由能台阶图,明确反应自发性,深化反应机理认知,为燃料电池、电解水等催化体系优化...
查看条目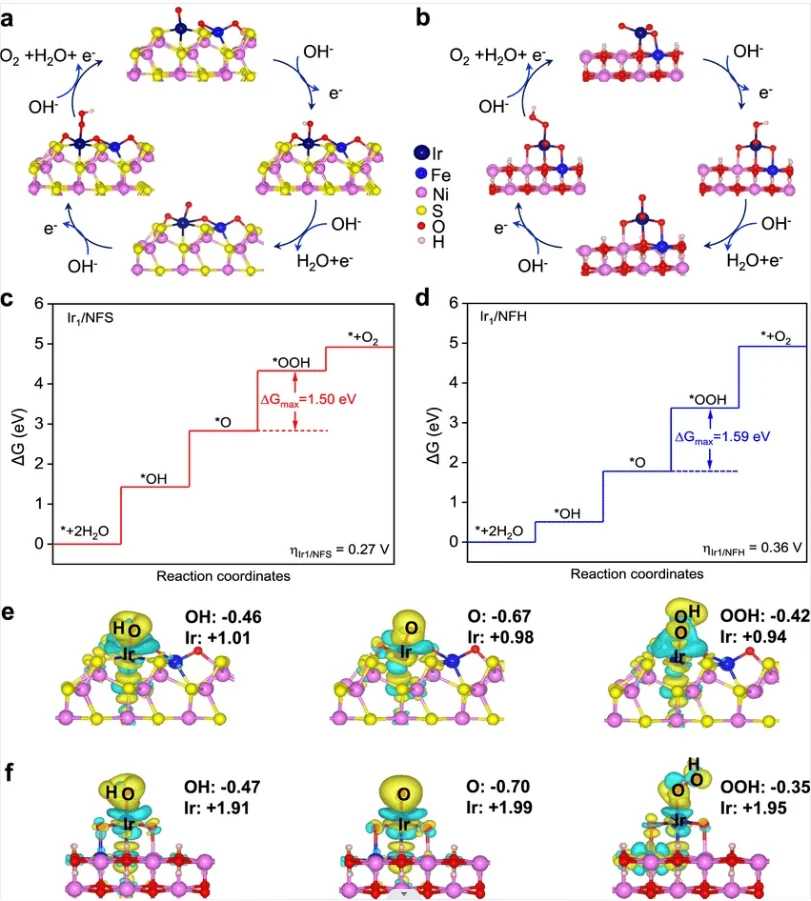
23
OER
OER(电催化析氧反应)
OER(电催化析氧反应)是能源转化领域核心四电子 - 质子耦合反应,涉及 * OH、*OOH 等氧中间体,存在晶格氧介导(LOM)、氧化物路径(OPM)及对称双活性位点 O-O 偶联等反应机制。基于 DFT 计算可精准解析反应机理,为电解水、燃料电池等领域高效析氧催化剂的设计与性能优化提供关键...
查看条目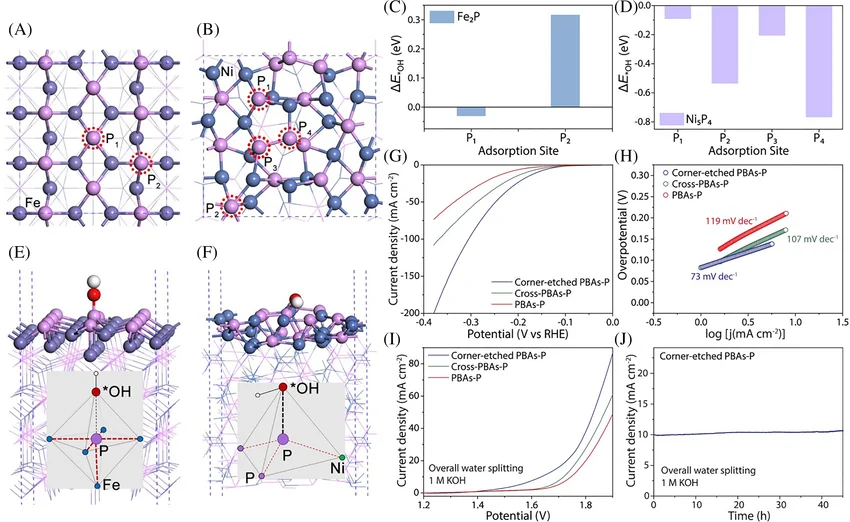
24
HER
HER(电催化析氢反应)
HER(电催化析氢反应)是能源转化领域核心反应,酸性与碱性条件下分别遵循 Volmer-Tafel、Volmer-Heyrovsky 两种反应机理。基于 DFT 计算可量化析氢步骤自由能差,解析反应机理,其中析氢步骤自由能越接近 0,催化活性越高,为高效析氢催化剂设计、能源转化体系优化提供关键...
查看条目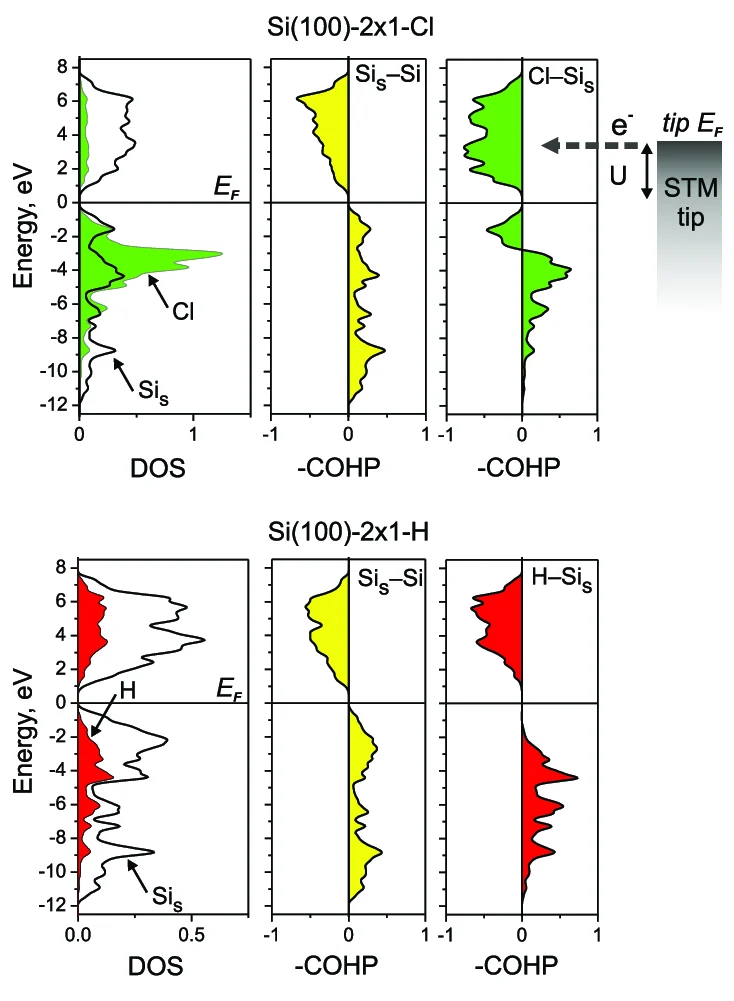
25
COHP
COHP(晶体轨道哈密顿布居)
COHP(晶体轨道哈密顿布居)是研究周期性体系局域化学键性质的核心工具,可直观表征成键与反键作用,常与 DOS(态密度)联合使用。其衍生参数 - pCOHP 中,>0 为成键区域、<0 为反键区域;费米能级向反键区域移动会导致材料失稳,向成键区域移动则提升材料稳定性,为周期性材料成键机理与稳定...
查看条目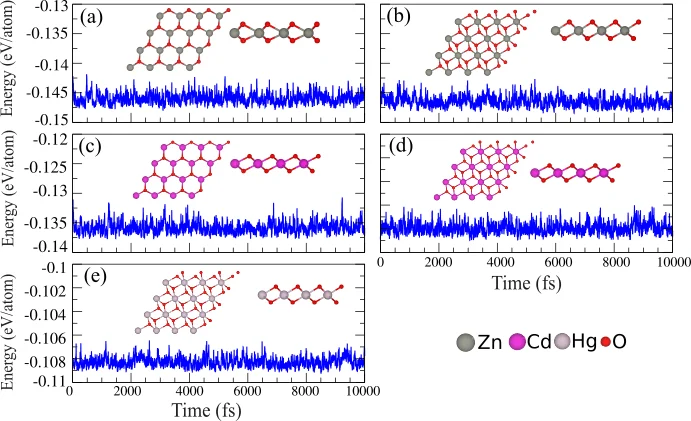
26
AIMD
AIMD(从头算分子动力学)
AIMD(从头算分子动力学,又称第一性原理分子动力学)是研究电子与原子核耦合系统力学演化的核心理论方法,被誉为原子分子尺度观测化学反应的 “显微镜”。无需预设力场参数,仅需原子初始结构,即可精准表征吸附能、扩散能垒及化学反应等特性,为微观动态过程解析与材料性能研究提供直接理论支撑。
查看条目围绕材料、催化、能源和电子结构研究,提供吸附能、能带/态密度、反应路径、电化学反应等第一性原理计算与结果分析。
FAQ
咨询前常见问题
这些问题用于帮助你整理任务条件,具体资源、周期和交付深度仍按项目确认。
第一性原理计算需要提供哪些输入? +
通常需要结构模型、研究目标、候选软件、计算参数偏好和希望输出的图谱或数据。若参数尚未确定,可以先说明问题背景,再共同确认计算口径。
DFT 图谱能否直接作为论文结论? +
图谱需要结合计算模型、参数收敛、对比组和研究问题解释。单张图通常不足以支撑完整结论,是否可用于论文表达需要按具体课题复核。
分子动力学模拟需要多长时间? +
模拟时间取决于体系规模、力场、时间步长、采样目标和资源条件。初期建议先确认代表性体系,再判断是否需要短程测试或长时间采样。
RMSD 平稳是否说明体系一定稳定? +
不一定。RMSD 是整体偏移指标,还需要结合轨迹、局部相互作用、能量变化和研究目标一起判断。单一指标不应替代体系复核。
分子对接结果是否需要再做 MD? +
是否需要取决于研究目标。如果只是快速筛选,对接可能足够;如果需要解释结合稳定性、构象变化或能量贡献,通常需要进一步模拟和复核。
有限元仿真前需要提供什么? +
建议提供几何模型、材料参数、载荷、约束、边界条件和希望观察的结果指标。如果工况不完整,可以先做建模可行性评估。