
典型任务
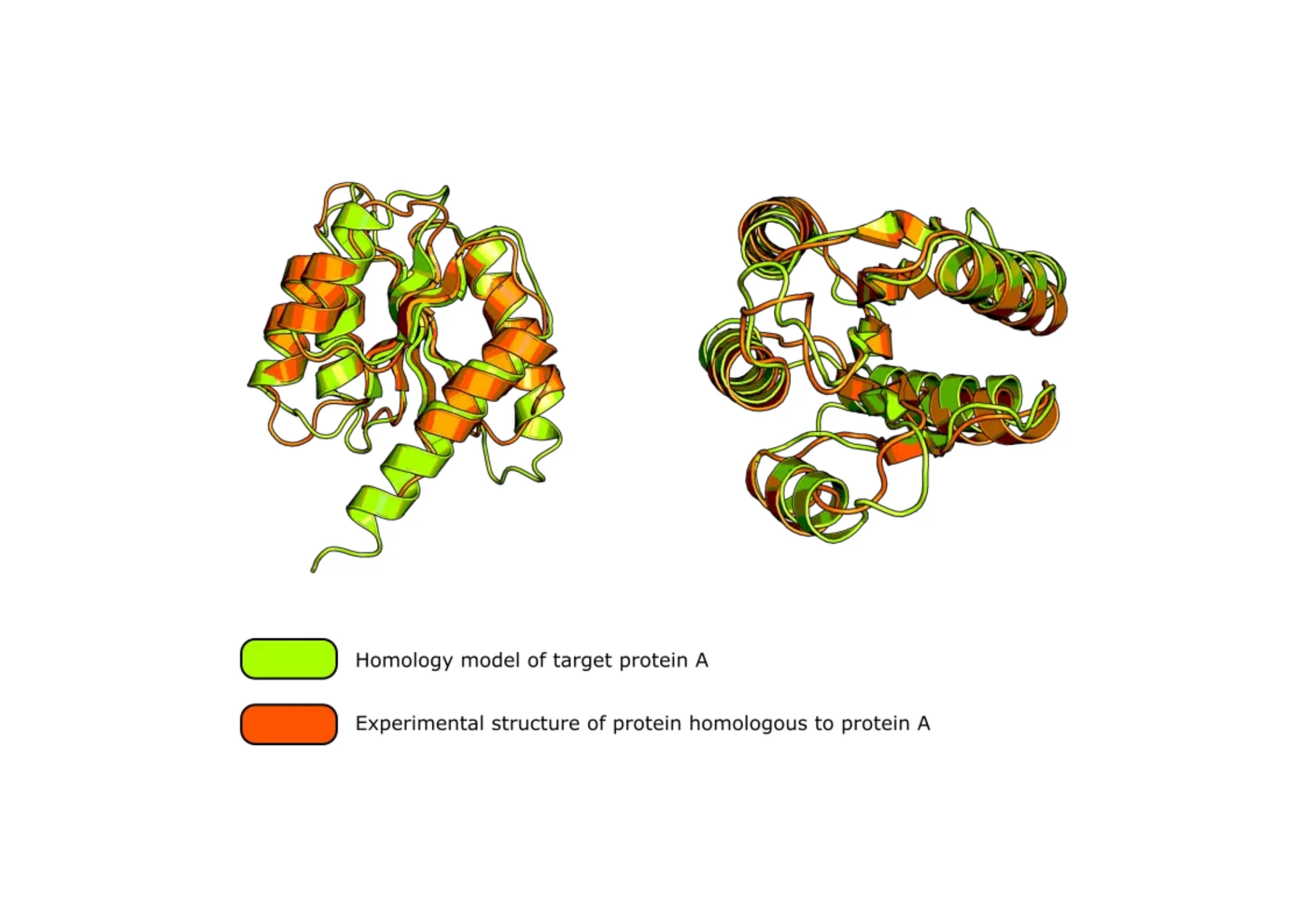
同源建模
RMSD(均方根偏差)
Rg(回旋半径)
氢键分析
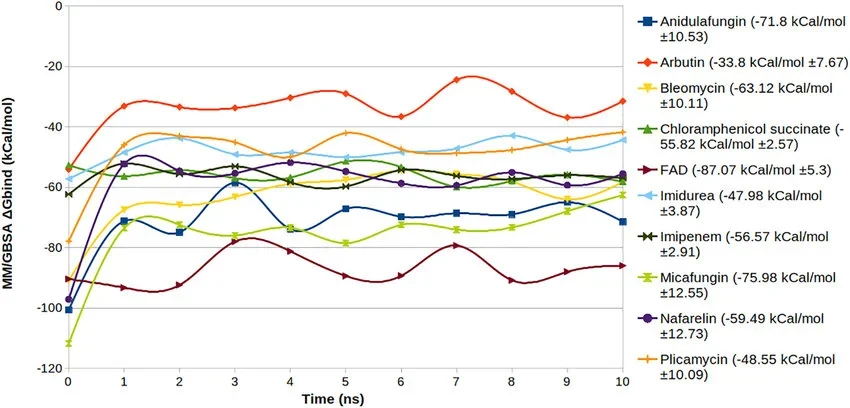
MM/GBSA
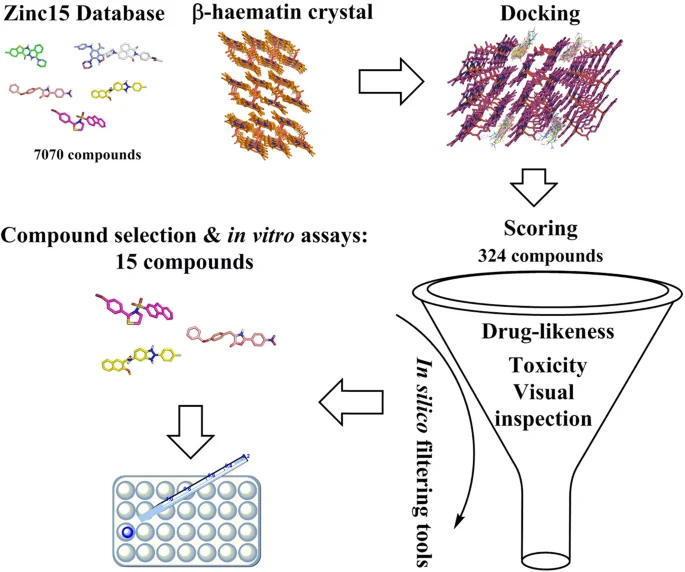
虚拟筛选
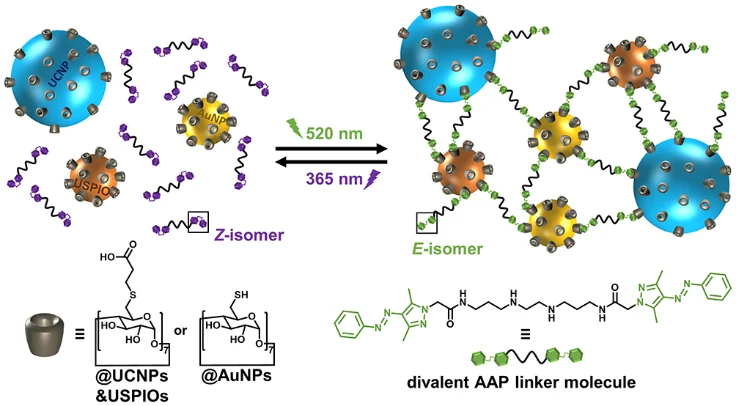
分子自组装
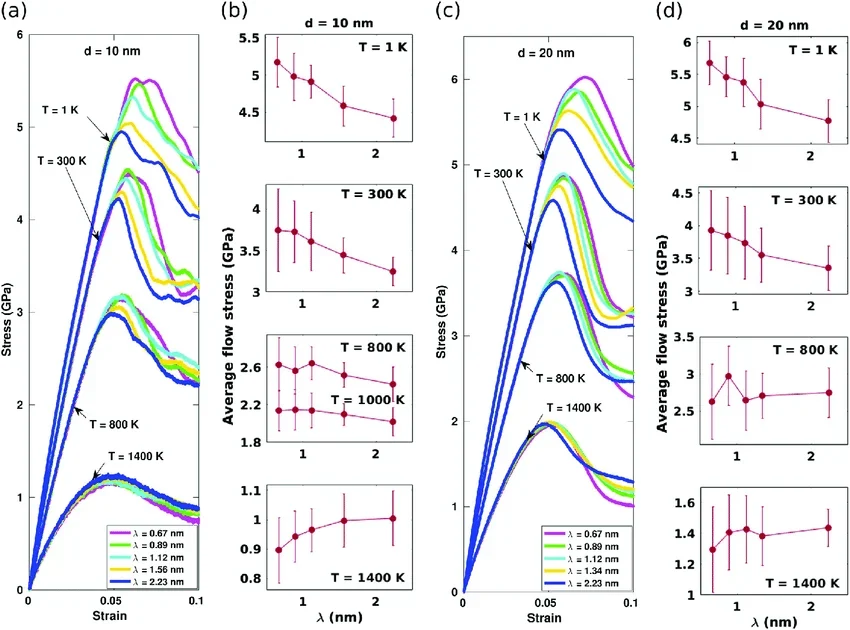
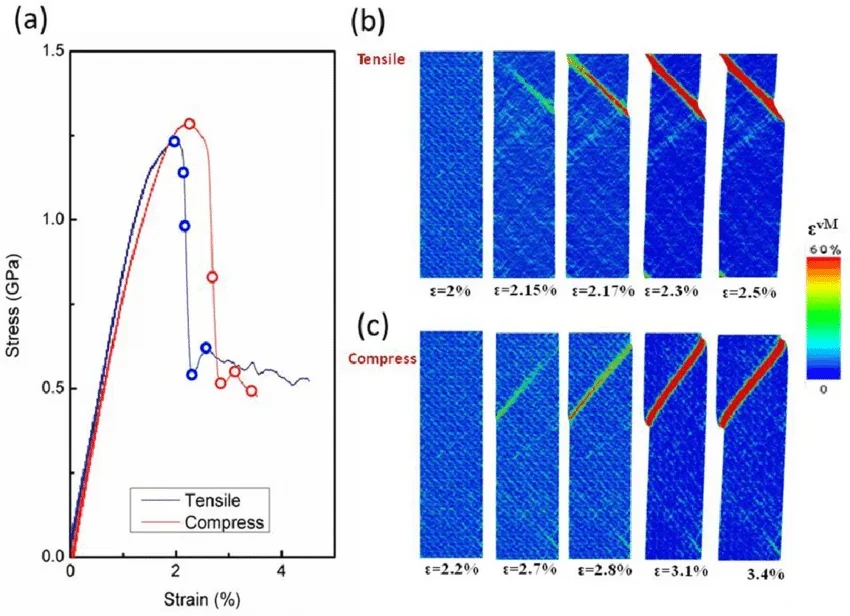
应力-应变曲线
分子动力学形变模拟
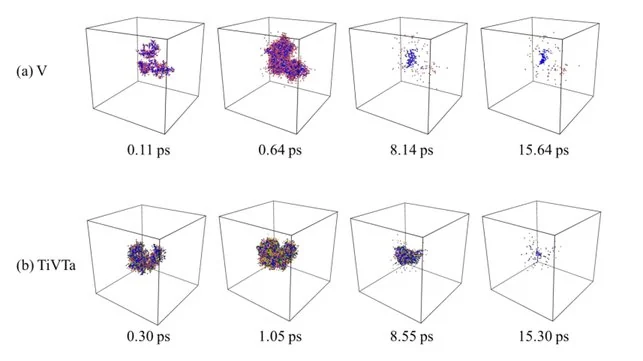
分子动力学辐照模拟
径向分布函数(RDF)
扩散系数
电池电解液的动力学模拟
热导率
栏目内容
按任务条目展开的计算内容
每个条目都有独立 URL、SEO 和搜索命中,用户可以按任务名称、分析指标和软件方向快速进入对应内容。
02
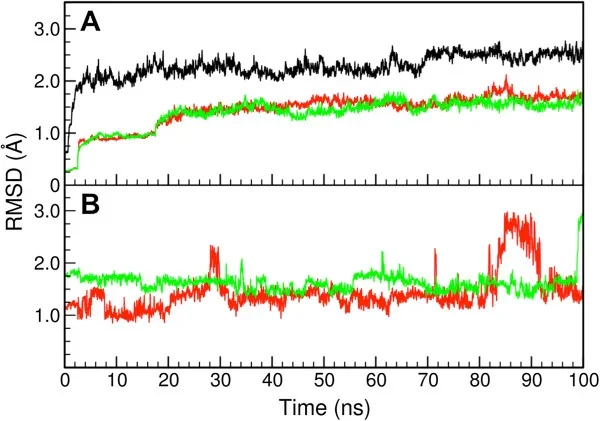
RMSD
RMSD(均方根偏差)
RMSD(均方根偏差)是分子动力学模拟中衡量结构变化的常用指标,通过计算模拟轨迹中各帧结构与参考结构(如初始构象)的原子位置偏差,来评估蛋白质等生物大分子的整体构象稳定性或折叠/去折叠过程。
查看条目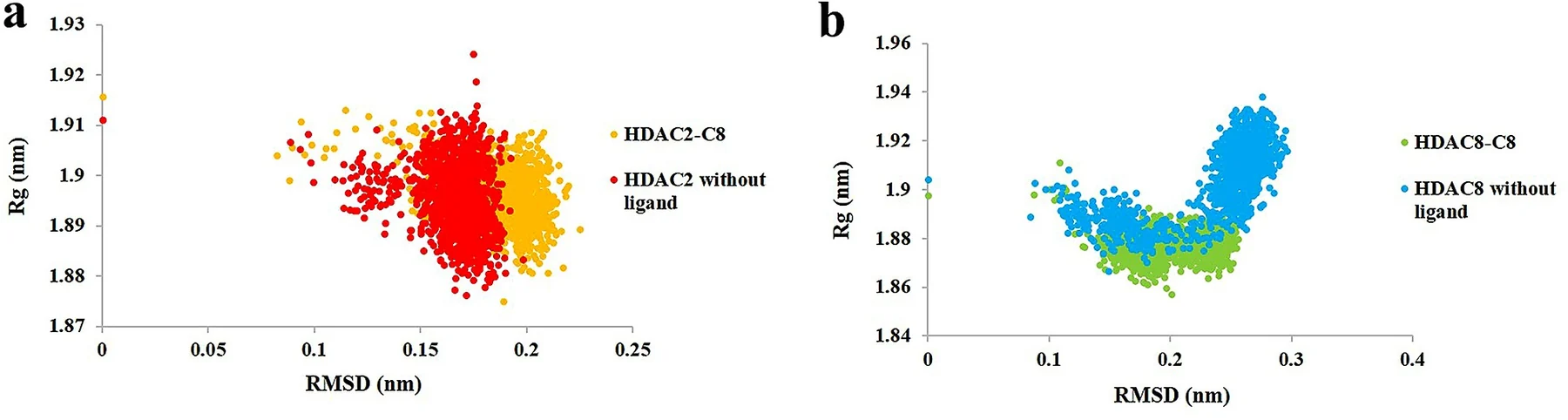
11
径向分布函数
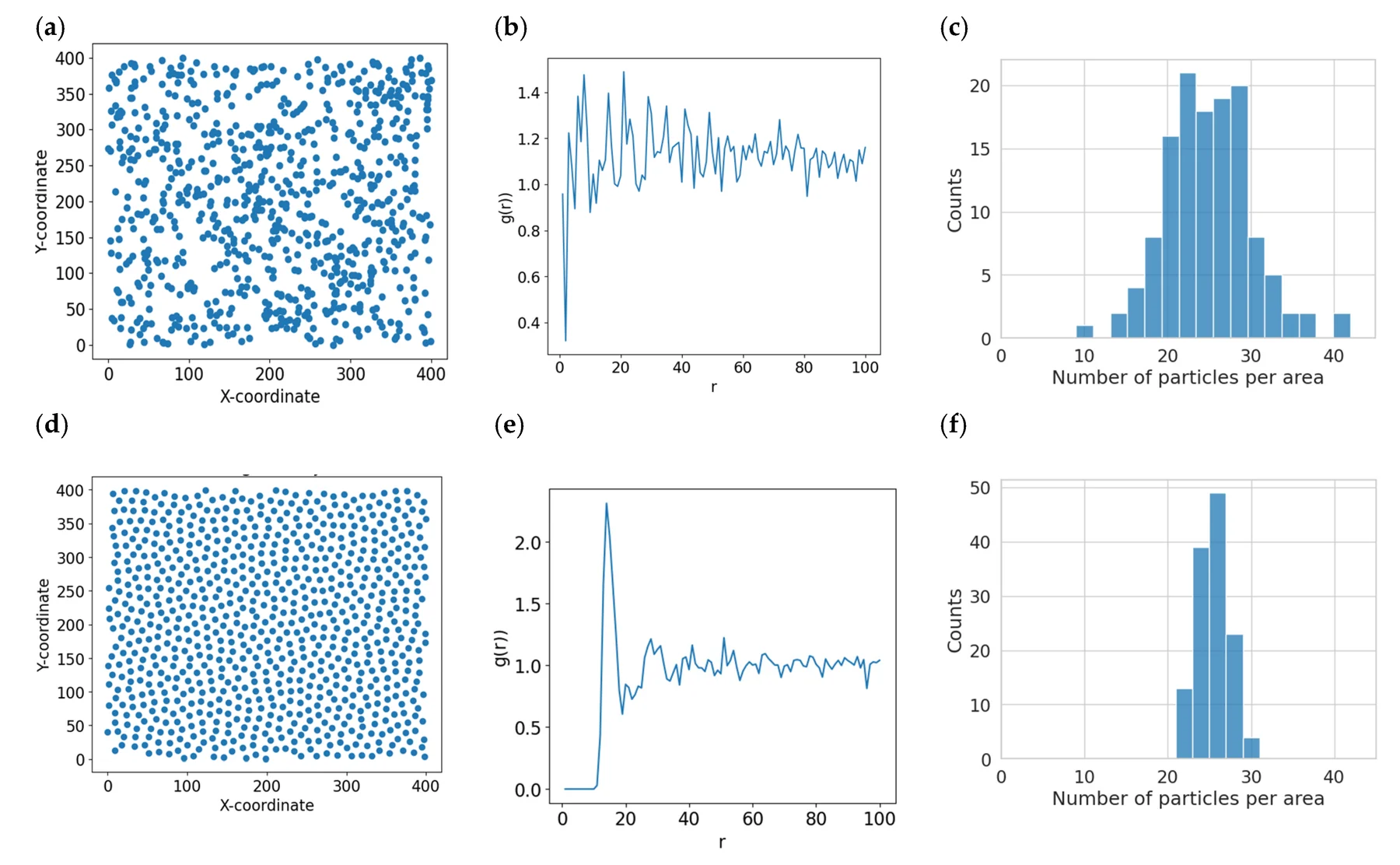
径向分布函数(RDF)
径向分布函数(RDF)用于表征模拟体系中粒子在空间的分布概率,通过统计以某一粒子为中心、不同距离处找到其他粒子的密度与体系平均密度之比,可分析溶液结构、离子水合、界面有序性等局部结构特征。
查看条目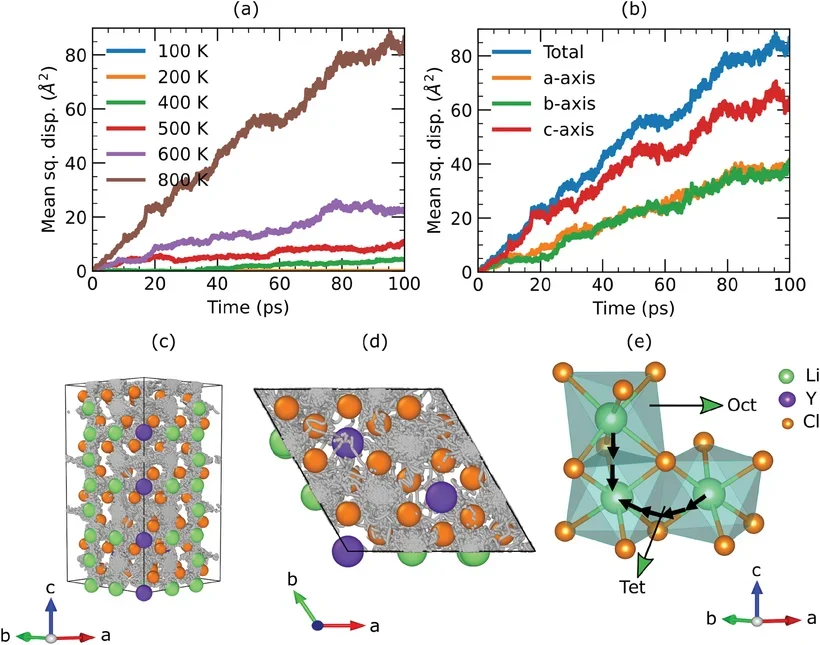
13
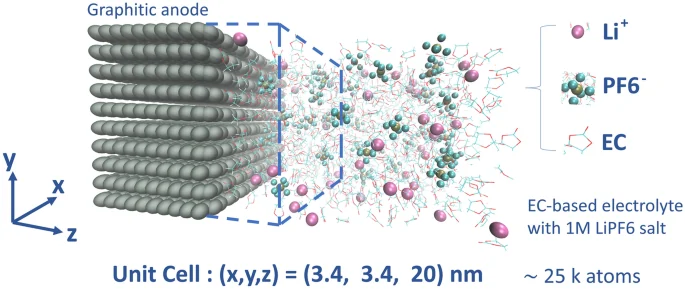
电池电解液的动力学模拟
电池电解液的动力学模拟
电池电解液的分子动力学模拟通过构建离子、溶剂分子与添加剂的原子模型,研究其微观结构(如离子溶剂化、团簇形成)、动力学性质(如扩散系数、电导率)及界面行为,为优化电解质配方与性能提供理论依据
查看条目
面向生物医药、材料和电池电解液等方向,支持同源建模、轨迹稳定性、结合自由能、扩散、热导率等分子动力学分析。
FAQ
咨询前常见问题
这些问题用于帮助你整理任务条件,具体资源、周期和交付深度仍按项目确认。
第一性原理计算需要提供哪些输入? +
通常需要结构模型、研究目标、候选软件、计算参数偏好和希望输出的图谱或数据。若参数尚未确定,可以先说明问题背景,再共同确认计算口径。
DFT 图谱能否直接作为论文结论? +
图谱需要结合计算模型、参数收敛、对比组和研究问题解释。单张图通常不足以支撑完整结论,是否可用于论文表达需要按具体课题复核。
分子动力学模拟需要多长时间? +
模拟时间取决于体系规模、力场、时间步长、采样目标和资源条件。初期建议先确认代表性体系,再判断是否需要短程测试或长时间采样。
RMSD 平稳是否说明体系一定稳定? +
不一定。RMSD 是整体偏移指标,还需要结合轨迹、局部相互作用、能量变化和研究目标一起判断。单一指标不应替代体系复核。
分子对接结果是否需要再做 MD? +
是否需要取决于研究目标。如果只是快速筛选,对接可能足够;如果需要解释结合稳定性、构象变化或能量贡献,通常需要进一步模拟和复核。
有限元仿真前需要提供什么? +
建议提供几何模型、材料参数、载荷、约束、边界条件和希望观察的结果指标。如果工况不完整,可以先做建模可行性评估。